Das Verhalten von Flüssigkeiten und Gasen wird in der Arbeitsgruppe von Prof. U. K. Deiters, Institut für Physikalische Chemie der Universität zu Köln, mit theoretischen Methoden untersucht. Das Ziel ist das Verständnis u.a. des Druck-Temperatur-Dichte-Verhaltens, der Verdampfungsenthalpie und der Wärmekapazität von Flüssigkeiten. Damit wollen wir langfristig in der Lage sein, zuverlässige Vorhersagen über die Eigenschaften von Flüssigkeiten zu machen, besonders in Bereichen, in denen Experimente problematisch, teuer oder gefährlich sind.
Mit der Schrödinger-Gleichung kann - im Bild der heutigen Wissenschaft - das Verhalten von Atomen und Molekülen grundsätzlich richtig wiedergegeben werden. Ein Wassertröpfchen auf der Haut enthält jedoch etwa 1025 Teilchen. Daher ist es aussichtslos, für ein derartiges System die Schrödinger-Gleichung aufzustellen und zu lösen. Deshalb werden zur Untersuchung Computersimulationen eingesetzt.
Simulationsverfahren zur Untersuchung von FlüssigkeitenComputersimulationen bieten sich an, um das Verhalten von derartigen Flüssigkeiten als Vielteilchensysteme zu untersuchen. Auf dem Gebiet der Flüssigkeiten konkurrieren zwei Simulationsverfahren: einerseits das Monte-Carlo-Verfahren (MC) und andererseits das Molekular-Dynamik-Verfahren (MD). Bei MC werden nacheinander zufällige Molekülanordnungen erzeugt und statistisch ausgewertet; MD hingegegen löst mit kleinen Zeitschritten die Bewegungsgleichungen der Moleküle. Beide Methoden liefern - nach einem Theorem der statistischen Thermodynamik - die gleichen Mittelwerte, haben aber jeweils ihre Vor- und Nachteile. In dieser Arbeit wird das MC-Verfahren verwendet. Ein Forscher, der auf dem Gebiet der Simulationen von Flüssigkeiten arbeitet, steht üblicherweise vor drei Problemen:
Mit diesen Fragen habe ich mich in meiner Doktorarbeit im Zeitraum 1996-1998 befaßt, in der die Eigenschaften von flüssigen Fluormethanen untersucht wurden. Seit dem Bekanntwerden der ozonschädigenden Wirkung der Fluor-Chlor-Kohlenwasserstoffe (FCKW) wurden weltweit erhebliche Anstrengungen unternommen, um den weiteren Abbau der Ozonschicht zu verhindern. So wurde mit dem Montrealer Abkommen der völlige Ausstieg bis zum Jahr 2000 aus der FCKW-Produktion beschlossen. Fluormethane bieten sich in diesem Zusammenhang als Ersatzstoffe an. Sie sind - ebenso wie FCKWs - ungiftig, unbrennbar, geruchs- und geschmackslos, haben jedoch keine ozonabbauende Wirkung. FCKWs wurden als Treibmittel, Lösungsmittel und in Kälteanlagen eingesetzt. Besonders im Bereich der Kältetechnik werden Fluor-Kohlenwasserstoffe als Ersatzstoffe eingesetzt.
Das Molekülmodell
Fluormethane bestehen aus fünf Atomen, bei denen die Wasserstoff- und Fluoratome annähernd tetraedisch um das zentrale Kohlenstoffatom angeordnet sind. Um diese zu modellieren, wird in dieser Arbeit ein starres Fünfzentren-Modell angenommen. Die Kraft zwischen den Molekülen wird somit auf die Wechselwirkung zwischen den gedachten Atomzentren zurückgeführt; bei zwei Molekülen mit 5 Atomen ergeben sich 25 Wechselwirkungen. Die Ladungsverteilung eines realen Moleküls ist jedoch nicht starr, sondern wird durch die umgebenden Moleküle verformt. Um diesen Einfluß zu berücksichtigen, wird im Molekülschwerpunkt eine Polarisierbarkeit angenommen. Damit bekommt jedes Molekül, in Abhängigkeit von der inviduellen Umgebung, ein zusätzliches Dipolmoment als vereinfachte Beschreibung der induzierten Ladungsverteilung.
Die ErgebnisseDie Simulationen geben die thermodynamischen, dielektrischen und strukturellen Eigenschaften der Verbindungen wieder. Der ganze Flüssigkeitsbereich zwischen Schmelzpunkt und kritischem Punkt kann mit einem polarisierbaren Teilchenmodell, das weder temperatur- noch druckabhängig ist, beschrieben werden.
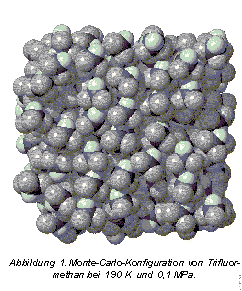
In Abbildung 1 ist eine MC-Konfiguration von Trifluormethan dargestellt. Sie gibt einen ersten Einblick in die Struktur der Flüssigkeit. Die Temperatur ist mit 190 K so eingestellt, daß sich die Flüssigkeit an ihrem Siedepunkt bei atmosphärischem Druck befindet. Das vorhandene Volumen wird von den Molekülen auffallend dicht ausgefüllt. Die Anordnung der Moleküle ist nicht, wie es auf den ersten Blick erscheint, regellos: Man erkennt die typische Nahordnung einer Flüssigkeit. Benachbarte Moleküle sind bevorzugt so orientiert, daß sich die entgegengesetzt geladenen Fluoratome (dunkel) und Wasserstoffatome (hell) möglichst nahe kommen.
In Abbildung 2 ist der Temperaturverlauf der Dichte von flüssigem Fluormethan dargestellt. Die simulierten und experimentellen Dichten stimmen über einen weiten Temperaturbereich gut überein. Neben der Dichte sind zahlreiche weitere Größen in den Simulationen berechnet worden. So konnte auch die Verdampfungsenthalpie bei den untersuchten Fluormethanen gut wiedergegeben werden. Diese Größe u.a. ist für den Bau von Kühlanlagen mit FCKW-Ersatzstoffen wichtig.
Need for Speed - Durchführung der Simulationen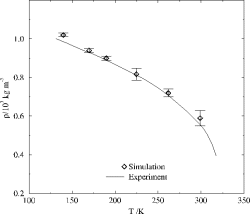
Simulationen mit polarisierbaren Flüssigkeitsmodellen benötigen extrem viel Rechenzeit. Jede Lageänderung eines Teilchens beeinflußt die induzierten Dipolmomente aller Moleküle. Die Iterationsroutine zur Berücksichtigung dieser Polarisationseffekte benötigt mit Abstand die meiste Rechenzeit des gesamten Programms.
Daher ist der Einsatz von leistungsfähigen Rechenanlagen und optimierten Algorithmen dringend notwendig. In dieser Arbeit wurden die Compute-Server des Regionalen Rechenzentrums der Universität zu Köln (RRZK) intensiv genutzt, insbesondere die leistungstarken Shared-Memory-Parallelrechner starfire (SUN, 40 Prozessoren) und sgi1 (Silicon Graphics, 16 Prozessoren). Für diese Anlagen wurden alle rechenintensiven Programmteile unter Verwendung von #pragma-Anweisungen parallelisiert. Während eines Programmlaufes wird über 95 Prozent der Rechenzeit in diesen parallelen Programmteilen verbracht. Durch Nutzung von vier Prozessoren konnte ein Geschwindigkeitszuwachs von 300 Prozent erreicht werden.
Weitere Rechnungen wurden auf dem Shared-Memory-Vektor-Parallelrechner Cray T90 am HLRZ in Jülich durchgeführt. Hierfür wurden die rechenintensiven Routinen so optimiert, daß die Vektorrecheneinheiten der Cray T90 optimal genutzt werden. Einzelne Routinen erreichen hierbei eine Geschwindigkeit von bis zu 980 MFLOPS (Millionen Fließkomma-Operationen pro Sekunde), das gesamte Programm erreicht 500-700 MFLOPS.
Neben diesen rein hardware-abhängigen Optimierungen wurden zahlreiche weitere Techniken untersucht und verwendet: so werden u.a. Mehrteilchenverschiebungen eingesetzt, damit bei jedem MC-Schritt eine möglichst große Änderung des Systems erreicht wird. Auf den beschriebenen Rechenanlagen wurden die Fluormethane CH3F, CH2F2 und CHF3 an mehreren Zustandspunkten untersucht. Für eine Rechnung bei einer typischen Flüssigkeitsdichte (Größenordnung 1 g/cm3) benötigen die Rechenanlagen am RRZK bis zu einem Monat Rechenzeit. Der größte Teil der Rechnungen wurde am RRZK durchgeführt. Darüberhinaus stellte das HLRZ in Jülich weitere 1.500 Stunden auf der Cray T90 für diese Arbeit zur Verfügung.
AusblickPolare Flüssigkeiten haben in Chemie, Biologie und Medizin eine zentrale Bedeutung. Sie werden als Lösungsmittel für organische Moleküle und Salze eingesetzt und bilden das Reaktionsmedium für die meisten chemischen Synthesen. Reaktionsverlauf und Geschwindigkeit werden durch die Wahl des Lösungsmittels gesteuert. Alle biochemischen Lebensprozesse laufen in einem wässerigen Medium ab. Diese Arbeit, auf dem Gebiet der reinen Fluide, ist ein erster Schritt zum Verständnis von Lösungsmitteleinflüssen. Die aufgestellten Modelle und entwickelten Simulationstechniken werden auf Mischungen und Lösungen übertragen. Es ist absehbar, daß zukünftig chemische und biochemische Prozesse in der flüssigen Phase modelliert werden.
back